




合作客戶/
拜耳公司 |
同濟大學 |
聯合大學 |
美國保潔 |
美國強生 |
瑞士羅氏 |






相關新聞Info
推薦新聞Info
-
> 基于粒徑、速度、表面張力、黏度測定揭示塵粒?霧滴碰撞行為規律(三)
> 基于粒徑、速度、表面張力、黏度測定揭示塵粒?霧滴碰撞行為規律(二)
> 基于粒徑、速度、表面張力、黏度測定揭示塵粒?霧滴碰撞行為規律(一)
> 高鹽低滲油藏中超低界面張力表面活性劑多段塞調驅機理與應用效果(三)
> 高鹽低滲油藏中超低界面張力表面活性劑多段塞調驅機理與應用效果(二)
> 高鹽低滲油藏中超低界面張力表面活性劑多段塞調驅機理與應用效果(一)
> 鈉鉀離子濃度對礦井水和純水表面張力、噴霧霧化特性的影響(三)
> 鈉鉀離子濃度對礦井水和純水表面張力、噴霧霧化特性的影響(二)
> 鈉鉀離子濃度對礦井水和純水表面張力、噴霧霧化特性的影響(一)
> Layzer模型與Zufiria模型研究界面張力對Rayleigh-Taylor氣泡不穩定性的影響
瀝青質及其亞組分與烷基苯磺酸鈉水溶液在降低IFT中的協同機理(二)
來源:高等學校化學學報 瀏覽 1219 次 發布時間:2024-09-27
2結果與討論
2.1瀝青質及其亞組分的元素分析
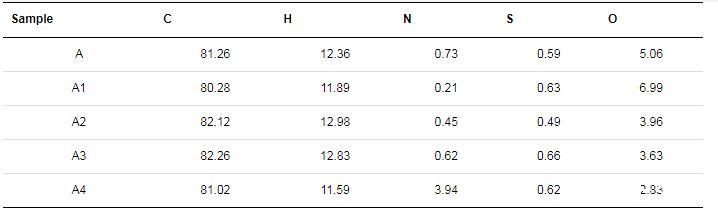
表1列出了瀝青質及其亞組分的元素組成分析結果,其中,O的元素組成通過差減法獲得。由表1可知,這些組分主要由C和H元素組成,雜原子(O,N,S)含量較低,與文獻報道的瀝青質元素分析結果一致。雜原子與碳原子和氫原子的質量比m(O)/m(C+H),m(N)/m(C+H)和m(S)/m(C+H)值(見圖2),可以反映瀝青質及其亞組分間雜原子比例的變化。比值越高,瀝青質及其亞組分的極性越高。由圖2可知,在瀝青質及其亞組分之間,m(O)/m(C+H)值變化明顯,其中,A1的m(O)/m(C+H)值最高,A和A2的m(O)/m(C+H)值高于A3和A4的;除A4的m(N)/m(C+H)值明顯較高外,其余m(N)/m(C+H)和m(S)/m(C+H)值變化均較小。由以上分析可知,A1的極性最高,而A3的極性最低。
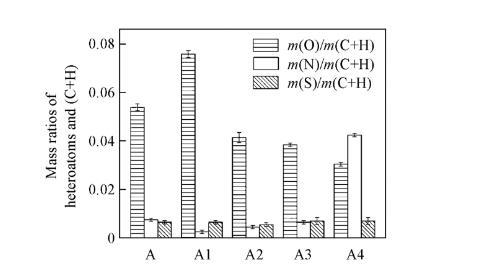
2.2瀝青質及其亞組分的FTIR分析
瀝青質及其亞組分的紅外光譜見圖3,由圖3可知,在3200——3400 cm-1范圍內均出現較寬的吸收峰,歸屬于締合的O—H和N—H的伸縮振動;在2922和2852 cm-1處均分別出現2個較強的吸收峰,分別歸屬于CH2的對稱和反對稱伸縮振動;在1640 cm-1處的吸收峰,由于1640和1600 cm-1間多個峰的延伸和重疊,因此很難給出此峰的準確歸屬;在1460和1380 cm-1處的吸收峰,歸屬于CH3的對稱和反對稱彎曲振動;在1040——1110 cm-1范圍內出現的吸收峰,歸屬于醚鍵的伸縮振動;在720 cm-1左右出現的吸附峰,歸屬于CH2的平面搖擺振動。
值得注意的是,所有組分的FTIR譜圖也存在明顯的區別:在3200——3400 cm-1范圍內,A3的吸收峰明顯弱于其它組分,表明A3的分子結構中締合的O—H和N—H含量較低,與A3的m(O)/m(C+H)與m(N)/m(C+H)值都較低的結果一致;在1710 cm-1處A,A3和A4均出現一個吸收峰,說明這些組分的分子結構中存在非締合的醛或羧酸(AR—COOH或R—COOH)或酮基;在1040——1110 cm-1范圍內,A1和A2的吸收峰強于A,A3和A4的,說明A1和A2的分子結構中含有較多的醚鍵;此外,在1040——1110 cm-1范圍內,與A,A3和A4相比,A1和A2的吸收峰紅移至1040 cm-1,這是由于醚鍵中的O與O—H或N—H基團之間形成氫鍵引起的,表明A1和A2具有較大的分子尺寸。
根據以上分析可知,與其它組分相比,A1和A2的分子結構中不僅極性雜原子“O”的含量高,而且具有較大的分子尺寸,這與元素分析的結果一致。
2.3瀝青質及其亞組分的平均相對分子質量
瀝青質及其亞組分的平均相對分子質量(M)的大小順序為A1(13495)>A2(6703)>A(2156)>A3(1735)>A4(1534)。A1和A2的M明顯大于A,A3和A4的,這表明A1和A2的分子尺寸較大,而A,A3和A4的分子尺寸大小相當,與瀝青質及其亞組分的紅外光譜分析結果一致。
2.4瀝青質及其亞組分的偶極矩
偶極矩可以用來度量瀝青質及其亞組分的極性大小。根據文獻中相應公式計算得到一系列含有不同濃度(質量分數)的瀝青質及其亞組分的甲苯溶液的介電常數。圖S1和圖S2(見本文支持信息)分別為瀝青質及其亞組分的介電常數和折射率隨濃度變化的曲線。通過外推法估算了各組分的介電常數和折射率,然后利用下式計算各組分的偶極矩:

式中:μ(C·m)為偶極矩;NA(6.022×1023mol-1)為阿伏伽德羅常數;n為折射率;ρ(1.2 g/cm3)為密度;k(1.381×10-23J/K)為波茲曼常數;T(K)為溫度;εr為介電常數;M為平均相對分子質量。
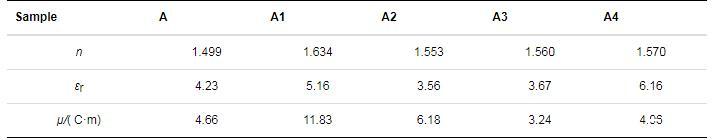
瀝青質及其亞組分的折射率、介電常數和偶極矩列于表2.由表2可知,A1的偶極矩最大,其次為A2,A4,A和A3.即A1的極性最大,A3的極性最小,這與瀝青質及其亞組分的元素分析和FTIR光譜分析結果一致。
2.5瀝青質及其亞組分對烷基苯磺酸鹽溶液與模擬油間動態界面張力的影響
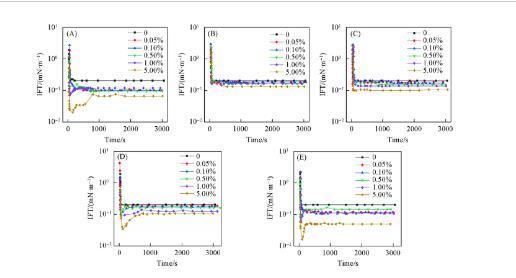
上述表征結果表明,瀝青質及其亞組分既有疏水的烷基鏈也有含雜原子(N,S和O)的親水基團,這一結構賦予了瀝青質及其亞組分一定的表面活性,能有效降低IFT.圖4為p-S14-4水溶液與含有不同濃度瀝青質及其亞組分的正十二烷模擬油DIFTs變化曲線。圖5為IFTmin隨瀝青質及其亞組分濃度變化的曲線。
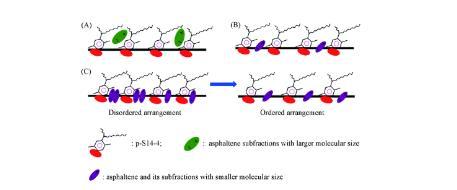
由圖4(B)和(C)可見,A1和A2的IFT持續下降直至平衡,與p-S14-4溶液和純的正十二烷模擬油體系的DIFTs的變化趨勢一致。DIFTs曲線呈“L”形,是最典型的DIFTs曲線類型之一。當油相與水相接觸時,p-S14-4分子迅速擴散并吸附到界面上,A1或A2由于分子尺寸較大向界面擴散速度平穩且緩慢,如圖6(A)所示。同時,界面上的活性物質開始脫附,但是吸附速率大于脫附速率,因此IFT持續下降。隨著時間的推移,吸附速率和脫附速率達到平衡,DIFTs達到平衡值IFTequ.在這種情況下IFTmin等于IFTequ.
從圖4(A),(D)和(E)可見,A,A3和A4的DIFTs變化趨勢與濃度密切相關。隨著瀝青質及其亞組分濃度的逐漸增加,DIFTs曲線由“L”型轉變為“V”型。這一有趣的結果是由活性分子在界面上呈現不同的排列方式引起的。當油相與水相接觸時,水相中的p-S14-4分子和油相中的界面活性分子都迅速擴散到界面并形成混合吸附膜。
最初,由于界面活性分子的持續吸附,且吸附速率大于脫附速率,導致IFT持續降低。在低濃度時,分子尺寸較小的瀝青質及其亞組分擴散驅動力小,難以突破油水界面能壘。
此外,活性分子稀疏地排列在界面上,如圖6(B)所示,并形成稀薄的混合吸附膜。因此,隨著時間的推移,吸附速率和脫附速率達到平衡,IFTs曲線也呈“L”形。在高濃度時,瀝青質組分獲得了較高的能量,能夠突破油水界面能壘,因此這些組分能夠更快地擴散到界面,并與P-S14-4分子無序地排列在界面上。當這些亞組分與P-S14-4分子之間形成了最緊密的混合吸附膜時,明顯的協同作用產生,導致IFT達到最小值。
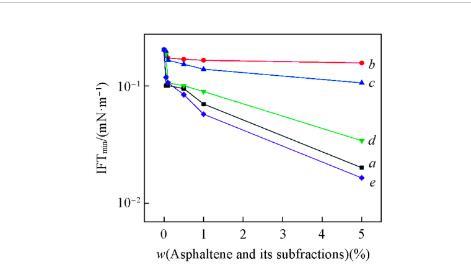
隨著時間的推移,界面上的活性分子由無序排列轉變為有序排列,如圖6(C)所示。在此過程中,界面活性分子的脫附速率大于吸附速率,界面濃度不斷減小,導致IFT升高直到脫附速率等于吸附速率。因此,DIFTs曲線呈“V”型。在此情況下,IFTmin不等于IFTequ.結果表明,小分子尺寸的瀝青質及其亞組分與P-S14-4分子的協同作用主要體現在瞬時的IFTmin值上。也可通過圖5證實,隨著A,A3和A4濃度的增加,IFTmin明顯降低,這表明隨著分子尺寸較小的瀝青質組分濃度的增加,混合吸附膜變緊密,協同作用增強。瀝青質主要通過與表面活性劑形成混合吸附膜和改變油相的性質來影響IFT。
從圖5可見,IFTmin隨A1和A2濃度的增加而減小。表明隨著A1和A2濃度的增加,油相的極性增加,油相和水相的性質差異減小。盡管A1和A2的極性較高,但IFTmin的變化不明顯,表明A1和A2的界面活性較弱。通常,分子極性越高,界面活性越強。而A1和A2表現出了不同的結果,主要與A1和A2有較大的分子尺寸有關。有較大分子尺寸的瀝青質亞組分,無論其極性如何都難以快速擴散并吸附到界面上,表明其界面活性較低。從圖5也可見,在同一低濃度時,A,A3和A4對IFTmin的影響幾乎相同;而在高濃度時,有較高極性的A和A4對IFTmin的影響明顯強于極性較低的A3.可見,有小分子尺寸的瀝青質組分能夠快速擴散并吸附到界面上,低濃度時,這些組分的界面濃度低,并且極性對IFTmin的影響較小;而在高濃度時,這些組分的界面濃度高,且極性越高,界面活性越強,越有利于縮小油水兩相的性質差異,導致IFTmin越低。
3結論
研究了瀝青質及其亞組分與p-S14-4分子在降低IFT中的協同作用。結果表明,瀝青質及其亞組分的分子尺寸、濃度和極性對協同作用有很大的影響。分子尺寸較大的瀝青質及其亞組分均難與p-S14-4分子形成混合吸附膜,因此,DIFTs曲線呈“L”型。在此情況下,IFTmin等于IFTequ.對于分子尺寸較小的瀝青質亞組分而言,在低濃度時,一方面,較低的擴散驅動力不足以使這些組分突破油水界面能壘;另一方面,這些亞組分與p-S14-4分子形成的混合吸附膜強度較弱,導致與p-S14-4分子的協同作用較弱。因此,DIFTs的變化不明顯,DIFTs曲線也呈“L”型。
在高濃度時,這些組分獲得了更高的能量去克服油水界面能壘。因此,這些組分能夠更快地擴散到界面,并與p-S14-4分子在界面上無序排列。當形成最緊密的混合吸附膜時,使IFT達到最小。隨著時間的延長,p-S14-4分子和這些組分由無序排列變為有序排列,導致IFT增加直至達平衡。此情況下,IFTmin不等于IFTequ,DIFTs曲線呈“V”型。IFTmin不僅與瀝青質組分的分子尺寸有關,還與極性有關。瀝青質及其亞組分的分子尺寸越小,向界面擴散的速度越快;極性越高,界面活性越強,越有利于縮小油水兩相性質差異。因此,既有較小的分子尺寸又有較高極性的瀝青質及其亞組分導致IFTmin變得更低。